PDEStrIAn has been published in a "perspective" in the ACS Journal of Medicinal Chemistry. Please refer to this paper when using the PDEStrIAn database or when refering to the in-depth structural analysis of PDE-inhibitors binding modes, the inhibitor binding mode classification, the binding site residue nomenclature, and the in-depth analysis and discussion regarding the large diversity of chemical scaffolds that are recognized by catalytic phosphodiesterase domains.
• How can I use PDEStrIAn?
For now, a good place to start with PDEStrIAn is via the quick-start tutorial that we developed for our kinase database KLIFS.
• What software and data sources are being used to create PDEStrIAn?
We are using in-house developed software (at the Division of Medicinal Chemistry, VU University Amsterdam) that combines many different software packages and multiple databases, but the heart of PDEStrIAn has been built using MOE from the Chemical Computing Group analyzing structural data from the Protein Data Bank.
The 3D-viewer (iview) is a WebGL viewer and requires no plugins to be installed. All up-to-date modern browsers support WebGL (more information), however, sometimes the configuration of the browser has to be adjusted. Please follow the steps described here to enable webGL for your specific browser.
• What is an MOE database?
MOE stands for Molecular Operating Environment and is a fully integrated drug discovery software package of the Chemical Computing Group. The MOE database download processes all the selected structures into a single fully annotated database file for this software package. See their website for more information: Chemical Computing Group
• What are these alternates/alternate models?
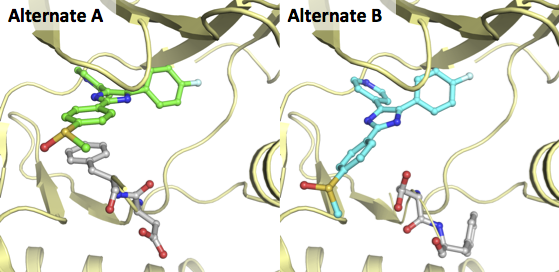
Alternates are alternative conformations or rotamers that can be defined for a single residue/substance/ligand in a crystal structure. A crystallographer can define alternates when the electron density for these elements are not concentrated on a single spot, but are clearly present on 2 or more spots. Each of the models will then be stored in an alternate (A, B, etc.). However, most PDB-viewer only load the first (A) model discards the rest of this information. As this information can be valuable we have chosen to split the alternates and store them in separate mol2 files.
A great example of two alternates for a ligand and residues is the X-ray of MAP kinase p38 in complex with a pyridinylimidazole (PDB-code 2EWA). In the figure below this structure is depicted for both alternates in exactly the same orientation:
• How can I download (all) processed structures from PDEStrIAn?
It is possible to download the processed PDE structures in MOL2 format or as a MOE database after performing a search using the download button at the bottom of the search results page. If you do not provide any search/filtering criteria PDEStrIAn will return all PDE entries within the database, therefore allowing you to download all structures from PDEStrIAn. All related annotations (IFP, pocket alignment, resolution, pockets, water clusters, etc.) are embedded within the MOE database or in a CSV file (overview.csv) within the MOL2 package.
• Can I combine different search options with each other?
Yes, all search options that are shown can be combined. So it is possible to search for an inhibitor that, for example, shares similarity with Sildenafil, belongs to the PDE4 subtypes, targets the pocket Q2, contains a Leucine at the HC2 position (residue 51) and has an H-bond interaction with the conserved gluatmine with a minimal crystal structure resolution of 2.5.
• I have identified an error (e.g. in the annotation, alignment, atom typing of a structure), can you correct it?
Please send us all information regarding the error using the Give us feedback button at the bottom of the webpage for this specific structure and we will correct it as soon as possible (or you can use the contact button at the bottom of this page).
• Can you please add a specific analysis or feature to your database/website?
Please contact us with your ideas for the improvement of PDEStrIAn (click on the contact button at the bottom).